以保护人民健康、促进创新发展为使命推动医疗器械临床评价改革向纵深化发展
2021年2月9日,国务院发布第739号令,公布新修订的《医疗器械监督管理条例》(以下简称新《条例》),该条例将于2021年6月1日起施行。新《条例》对医疗器械临床评价相关要求进行了全面修订,包括免于进行临床评价的情形(第二十四条)、临床评价路径和应当开展临床试验的医疗器械(第二十五条)、鼓励和支持医疗机构开展临床试验(第二十六条),临床试验审批的默示许可(第二十七条),临床试验的伦理审查和知情同意(第二十八条)、临床急需医疗器械的拓展使用(第二十九条)等方面。
本文从新《条例》医疗器械临床评价相关要求的修订背景、修订内容、现实意义以及落实措施展开,阐明新《条例》如何以法规形式巩固审评审批制度的改革成果,并从制度层面推动医疗器械临床评价改革向纵深化发展,进一步促进产业创新发展,以更好满足人民对高质量高性能医疗器械的需求。
修订背景:审评审批制度的改革成果和中国主导制定的国际协调文件
(一)巩固药品医疗器械审评审批制度的改革成果
2017年10月,中共中央办公厅、国务院办公厅印发《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,提出改革临床试验管理的要求。
经过三年的改革实践,在临床评价规范性文件的制定和具体产品的审评方面,取得了阶段性成果,有效促进了创新医疗器械的加快上市,以及临床急需医疗器械的患者可及。《医疗器械拓展性临床试验管理规定(试行)》《真实世界数据用于医疗器械临床评价技术指导原则(试行)》《接受医疗器械境外临床试验数据技术指导原则》《医疗器械临床试验设计指导原则》等文件的发布和实施,丰富和拓宽了临床数据的来源,解决了如何开展临床试验的问题,产生了临床评价的新方法和新工具,形成了科学的临床评价思路。在具体产品的审评审批工作中,各类产品的临床评价路径在监管机构和业界基本达成共识,产品注册和许可事项变更项目中临床试验比例得到有效降低,处于合理水平。
(二)落实中国主导制定的临床评价国际协调文件
2018年3月,由国家药品监督管理局提出的“医疗器械临床评价(MDCE)”新项目建议,在医疗器械国际监管者论坛(IMDRF)第13次IMDRF管理委员会会议上成功立项。2019年9月,由中国监管机构牵头,国际主要监管机构参与制定的项目成果文件《临床证据-主要定义和概念》《临床评价》《临床试验》在第16次IMDRF管理委员会会议上顺利通过,作为医疗器械临床评价领域的国际协调文件在IMDRF官网上正式发布。
三个国际协调文件全面、系统地阐明了医疗器械临床评价领域的相关问题,包括主要定义和概念,临床评价的作用、范围、流程、路径和数据来源,何时需要以及如何开展临床试验等。上述文件既是国际上医疗器械临床评价新理念、新思路和新方法的有效反映,更是中国审评审批制度改革成果的集中体现。中国作为IMDRF成员国,作为上述协调文件的牵头起草者,需按照IMDRF标准操作规程,实施已发布的协调文件。
修订内容:建立与产品特征和临床风险相适应的、明晰的临床评价要求
通过全面梳理、深入分析和精准提炼审评审批制度的改革成果和国际协调文件的相关要求,新《条例》以高度概括的语言对临床评价相关条款进行了修订。
(一)首次提出“可以免于进行临床评价的情形”
第二十四条提出“医疗器械产品注册、备案,应当进行临床评价,但是符合下列情形之一,可以免于进行临床评价:
工作机理明确、设计定型,生产工艺成熟,已上市的同品种医疗器械临床应用多年且无严重不良事件记录,不改变常规用途的;
其他通过非临床评价能够证明该医疗器械安全、有效的”。
(二)明晰表述“进行医疗器械临床评价的路径”,准确界定“应当开展临床试验的医疗器械”
第二十五条明确“进行医疗器械临床评价,可以根据产品设计特征、临床风险、已有临床数据等情形,通过开展临床试验,或者通过对同品种医疗器械临床文献资料、临床数据进行分析评价,证明医疗器械安全、有效”。
“按照国务院药品监督管理部门的规定,进行医疗器械临床评价时,已有临床文献资料、临床数据不足以确认产品安全、有效的医疗器械,应当开展临床试验”。
(三)在强化临床试验质量管理的基础上,支持和鼓励医疗机构开展医疗器械临床试验
在规范临床试验质量管理、临床试验备案和临床试验机构备案的基础上,第二十六条进一步提出“国家支持医疗机构开展临床试验,将临床试验条件和能力评价纳入医疗机构等级评审,鼓励医疗机构开展创新医疗器械临床试验”。第二十八条强调开展医疗器械临床试验,应当进行伦理审查,获得受试者知情同意,不得收取与临床试验有关的费用。
(四)高风险临床试验审批实行“默示许可”
第三类医疗器械临床试验对人体具有较高风险的,应当经国务院药品监督管理部门批准。第二十七条明确指出,国务院药品监督管理部门审批临床试验,“自受理申请之日起60个工作日内作出决定并通知临床试验申办者。逾期未通知的,视为同意”。
(五)拓展使用“治疗严重危及生命且尚无有效治疗手段的疾病的医疗器械”
第二十九条提出,“对正在开展临床试验的用于严重危及生命且尚无有效治疗手段的疾病的医疗器械,经医学观察可能使患者获益,经伦理审查、知情同意后,可以在开展医疗器械临床试验的机构内免费用于其他病情相同的患者,其安全性数据可以用于医疗器械注册申请”。
修订意义:进一步保障产品安全有效,促进产业创新发展
(一)严守产品安全有效底线,保护人民生命安全和身体健康
以医疗器械安全有效为目标,综合考虑产品设计特征、临床风险、应用历史等因素,设置与之相适应的临床评价要求。对于非临床评价能够证明产品安全、有效的,免于进行临床评价;进行临床评价的,可通过对同品种医疗器械临床文献资料、临床数据进行分析评价,证明产品安全有效;当已有临床文献资料、临床数据不足以确认产品安全、有效的,应当开展临床试验。分层次的临床评价要求,与具体产品的安全有效性评价相适应,且临床评价目标明确、重点突出,有利于严守安全有效底线,保护人民生命安全和身体健康。
(二)进一步明确临床评价要求,合理降低行业负担,促进产业创新发展
新《条例》明确指出临床评价的路径和相关要求,即同品种临床评价路径和临床试验路径。对于是否开展临床试验,从现行《条例》“申请第二类、第三类医疗器械产品注册,应当进行临床试验”到新《条例》“进行医疗器械临床评价时,不足以确认产品安全、有效的医疗器械,应当开展临床试验”,确定了根据产品安全有效性评价的需要,决定是否开展临床试验的原则,大大缩小了开展临床试验的范围,与国际协调文件的临床试验决策思路一致,合理降低了行业负担,促进产业创新发展。
(三)鼓励和支持医疗机构开展临床试验,为临床评价提供高质量数据
新《条例》明确指出,国家支持和鼓励医疗机构开展临床试验,并将临床试验条件和能力评价纳入医疗机构等级评审,为医疗机构积极开展医疗器械临床试验、提高临床试验的能力和水平,注入了驱动力,有利于提高临床试验质量,为医疗器械临床评价提供高质量的数据。
(四)遵循科学原则,强调规范管理,拓展国际视野,推动医疗器械临床评价改革向纵深化发展
基于产品安全有效性评价的需要,设置不同的临床评价要求,决策是否开展临床试验,遵循了临床评价的科学原则;药品监督管理部门制定相关指南及目录,强调了临床评价的规范化管理以及相关要求的公开透明;临床评价的整体要求,与IMDRF国际协调文件高度一致,体现了国际视野,有利于申请人的临床评价文件在不同国家和地区间的互通互用。
落实措施:构建医疗器械临床评价通用指导原则体系,有效指导临床评价工作的开展
为贯彻落实新《条例》中临床评价的相关要求,将发布系列配套的临床评价通用技术指导原则,包括《医疗器械临床评价技术指导原则(修订)》《医疗器械临床评价报告撰写技术指导原则》《决策是否开展医疗器械临床试验技术指导原则》《医疗器械等同性论证技术指导原则》《体外诊断试剂临床试验技术指导原则(修订)》等。上述指导原则和审评审批制度改革中发布临床评价相关技术指导原则一起,初步构建了医疗器械临床评价通用指导原则体系(见表),有效指导监管机构和行业开展相关工作。
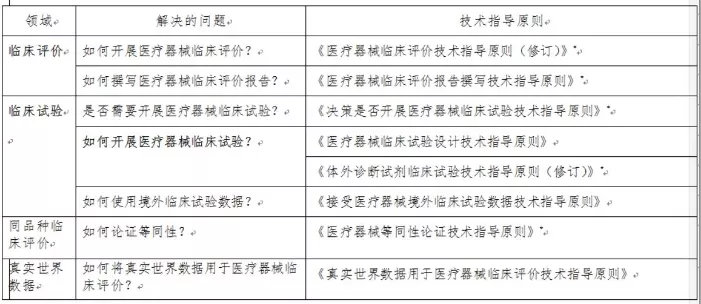
来源:中国器审
;){kind=link}